
So what is a FASTA file, anyway?
I remember that when I first started working in a bioinformatics lab, the more experienced lab members were always talking about “fast-ə” (“fast-eh”) and “fast-q” files and I, for longer than I care to admit, remained uncertain of exactly what they were.
In bioinformatics, FASTA files are a type of plain text file used to link metadata with other information, often biological sequence data. Typically, FASTA files end in .fa, .fna or .fasta and use the > character as a delimiter.
FASTA files are relatively simple. The delimiter is an indicator that a new entry has begun; everything from the current delimiter to the next represents data that is related in some way. Lines that begin with a delimiter are loosely termed “headers”, which usually alternate with lines containing other data. Because most computational processing involves moving through files sequentially, this format allows the interpreting software to “ask” itself on each line “Have I reached another delimiter yet?” If the answer is “no”, the line is inferred to be associated with the previous header.
Aside Because genome sizes are measured in number of bases (along with the standard unit prefixes, e.g. kilo, mega, giga), and because each text character is ~1 byte, there is a nice correspondence between genome size in bases and plain text genome size on disk. For example, the human genome comprises ~3.23 gigabases (or Gb), and it’s file size is ~3.15 gigabytes (GB):
3151425857 Jan 16 2015 Homo_sapiens.GRCh38.dna.primary_assembly.faFASTA format sample
A FASTA-formatted genome file might have entries like this for sequence data that has been successfully assembled into chromosomes:
>Chromosome_1 [optional metadata here]
GATGTGACTATATACTTAGGTTCGATCTCGTCCCGAGAATTTTAAGCCTCAGCATCTACGAGTTATGAGGTTAGCCAAA
AAAGCACGTGGTGGCGCCCACCGACTGTTCCCAAACTGTAGCTCTTCGTTCCGTCAAGGCCCGACTTTCATCGCGGCCC
ATTCCATGCGCGGACCATACCGTCCTAATTCTTCGGTTATGTTTCCGATGTAGGAGTGAGCCTACCTGCCTTTGCGTCT
[sequence continued...]
>Chromosome_2 [optional metadata here]
GCGGGGCTCTTGACGTTTGGAGTTGTAAATATCTAATATTCCAATCGGCTTTTACGTGCACCACCGCGGGCGGCTGACG
AGGGACTCACACCGAGAAACTAGACAGTTGCGCGCTGGAAGTAGCGCCGGCTAAGAAAGACGCCTGGTACAGCAGGACT
ATGAAACCCGTACAAAGGCAACATCCTCACTTCGGTGAATCGAAACGCGGCATCAAGGTTACTTTTTGGATACCTGAAA
[sequence continued...]There’s a lesson in them there newlines
One of the mantras my PI emphasized early on is, always try to “touch” the data as much as possible. Assuming you’re running a Unix-based OS, this means getting comfortable (if you aren’t already) with basic file commands like more, less, head, tail, etc. The field of bioinformatics is still replete with standardization issues, and getting into the habit of moreing files as soon as you download them just to make sure you know what you’re working with is a good idea.
For an example of where this can be useful, suppose I have just downloaded a new genome and I want to do some sort of parsing of it. What I’d like to do is write a small module to allow me to associate headers and their sequences, which could be used to do further downstream work inside of a larger script. No beginner bioinformatician, I first locate the file in my terminal and more it to make sure everything is in order:
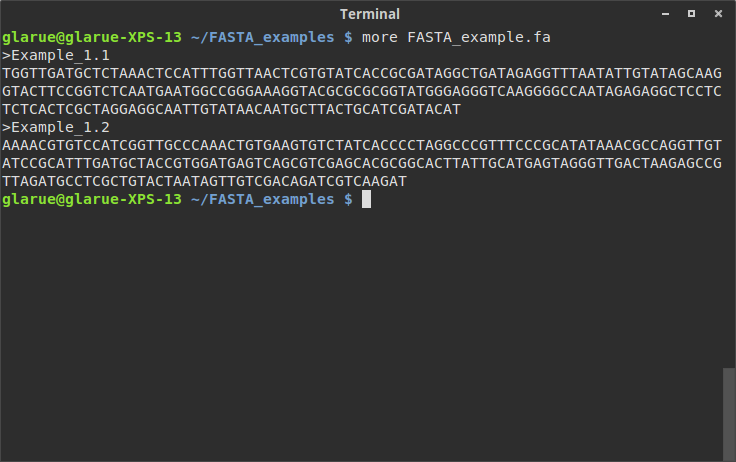
Looks like a standard FASTA file so far. But wait, are those sequences split over multiple lines? One way to check this is just to maximize the width of the terminal window (the first more is with the terminal at default size, the second is after it’s been widened):
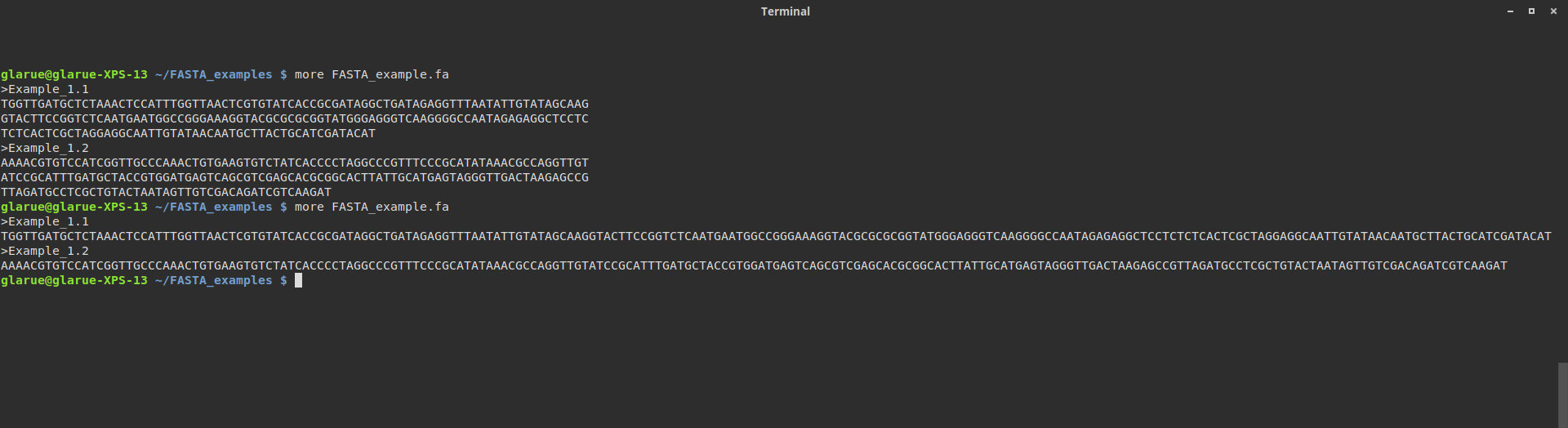
Okay, it looks like each header is followed by a single line containing all of the sequence. Line breaks aren’t something most people need to think about, but if you’re going to be writing code to parse text from files, accounting for line breaks comes with the territory.
So, now that I understand the file format I can write a short Python script to pull out the headers and the associated sequences. Since each content line follows its header, and the file starts with a header, I’ll just assign each pair of lines to a corresponding pair of header/sequence variables:
def easy_fasta_parse(fasta):
header, seq = None, None # start the file with empty variables
with open(fasta) as f:
for line in f:
if not header: # first line assigned to header
header = line.lstrip(">").strip() # remove delimiter and newline
continue
else: # we already have one stored in header
current_header = header # allow for reset of header variable
header = None # reset header
seq = line.strip()
yield current_header, seq # offer up header and sequenceLet’s see how the above script does on our test file:
for h, s in easy_fasta_parse("FASTA_example_2.fa"):
print("HEADER:", h)
print("SEQ:", s)HEADER: Example_1.1
SEQ: TGGTTGATGCTCTAAACTCCATTTGGTTAACTCGTGTATCACCGCGATAGGCTGATAGAGGTTTAATATTGTATAGCAAGGTACTTCCGGTCTCAATGAATGGCCGGGAAAGGTACGCGCGCGGTATGGGAGGGTCAAGGGGCCAATAGAGAGGCTCCTCTCTCACTCGCTAGGAGGCAATTGTATAACAATGCTTACTGCATCGATACAT
HEADER: Example_1.2
SEQ: AAAACGTGTCCATCGGTTGCCCAAACTGTGAAGTGTCTATCACCCCTAGGCCCGTTTCCCGCATATAAACGCCAGGTTGTATCCGCATTTGATGCTACCGTGGATGAGTCAGCGTCGAGCACGCGGCACTTATTGCATGAGTAGGGTTGACTAAGAGCCGTTAGATGCCTCGCTGTACTAATAGTTGTCGACAGATCGTCAAGATGreat, that worked exactly like we wanted; the script correctly parsed each header and sequence line, and alternated between them successfully. Now let’s say we grab another genome that we’d like to look at. more shows a structure identical to the first file:
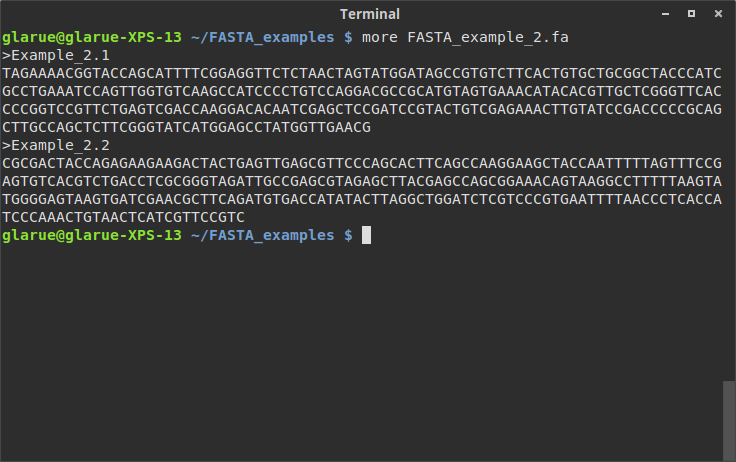
Let’s run our little script on it:
for h, s in easy_fasta_parse("FASTA_example_2.fa"):
print("HEADER:", h)
print("SEQ:", s)HEADER: Example_2.1
SEQ: TAGAAAACGGTACCAGCATTTTCGGAGGTTCTCTAACTAGTATGGATAGCCGTGTCTTCACTGTGCTGCGGCTACCCATC
HEADER: GCCTGAAATCCAGTTGGTGTCAAGCCATCCCCTGTCCAGGACGCCGCATGTAGTGAAACATACACGTTGCTCGGGTTCAC
SEQ: CCCGGTCCGTTCTGAGTCGACCAAGGACACAATCGAGCTCCGATCCGTACTGTCGAGAAACTTGTATCCGACCCCCGCAG
HEADER: CTTGCCAGCTCTTCGGGTATCATGGAGCCTATGGTTGAACG
SEQ: >Example_2.2
HEADER: CGCGACTACCAGAGAAGAAGACTACTGAGTTGAGCGTTCCCAGCACTTCAGCCAAGGAAGCTACCAATTTTTAGTTTCCG
SEQ: AGTGTCACGTCTGACCTCGCGGGTAGATTGCCGAGCGTAGAGCTTACGAGCCAGCGGAAACAGTAAGGCCTTTTTAAGTA
HEADER: TGGGGAGTAAGTGATCGAACGCTTCAGATGTGACCATATACTTAGGCTGGATCTCGTCCCGTGAATTTTAACCCTCACCA
SEQ: TCCCAAACTGTAACTCATCGTTCCGTCHmmm, that didn’t work. What went wrong? Oh, right, that whole lack-of-format-standardization thing! Let’s check the file again, resizing the terminal as before to check the line breaks:
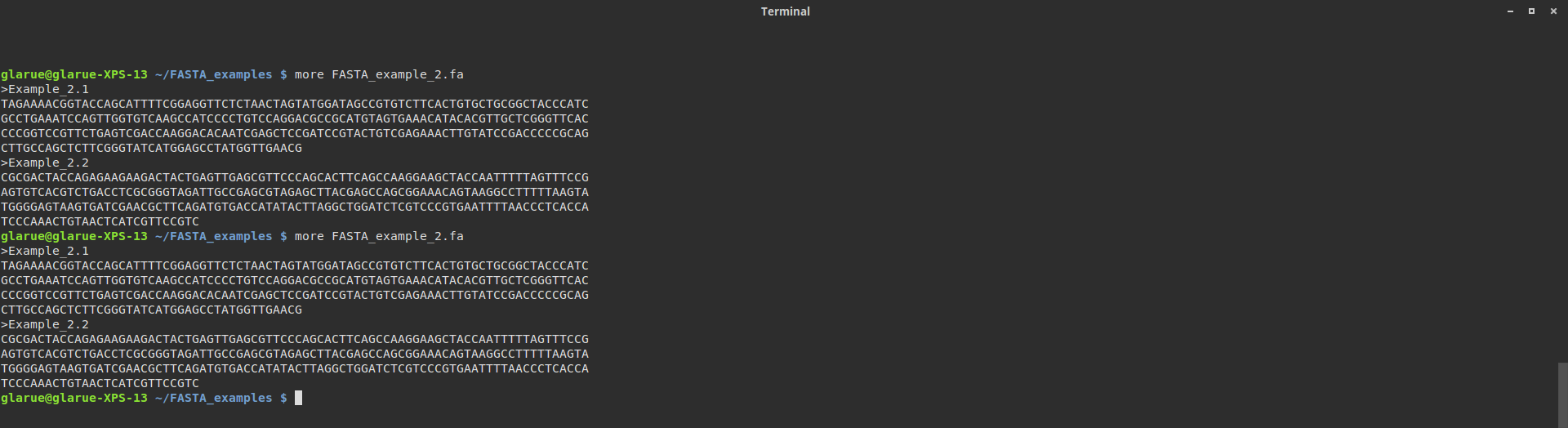
Unlike in the first example, the line formatting didn’t change. That is because in this particular FASTA file, each line is <= 80 characters long. Do not ask me why anyone would choose to format their FASTA file in such a way - I know only that people have (perhaps simply to spite me, who knows?) and will probably continue to do so.
This type of issue is a good motivator for making one’s code as general as possible, as often as possible. For every project, of course, one must weigh the time investment of writing a more robust, general solution versus something quick and dirty that just needs to work once. Sometimes you really know that you’re only going to do something once, so “works for this specific file” is good enough. In this case, however, a mechanism for retrieving the headers and corresponding sequences from a FASTA file is likely to come in useful in many different applications, and it’s therefore probably worth doing it more carefully. Here, I’ve written a more general parser which provides greater flexibility in its handling of both input and output:
def fasta_parse(fasta, delimiter=">", separator="", trim_header=True):
"""
Iterator which takes FASTA as input. Yields
header/value pairs. Separator will be
used to join the return value; use separator=
None to return a list.
If trim_header, parser will return the
FASTA header up to the first space character.
Otherwise, it will return the full, unaltered
header string.
"""
header, seq = None, []
with open(fasta) as f:
for line in f:
if line.startswith(delimiter):
if header is not None:
if separator is not None:
seq = separator.join(str(e) for e in seq)
yield header, seq
header = line.strip().lstrip(delimiter)
if trim_header:
try:
header = header.split()[0]
except IndexError: # blank header
header = ""
seq = []
elif line.startswith('#'):
continue
else:
if line.strip(): # don't collect blank lines
seq.append(line.rstrip('\n'))
if separator is not None:
seq = separator.join(str(e) for e in seq)
yield header, seqThere are certainly other - and likely better - ways of approaching the same problem, and the extra flexibility I’ve included in the above parser is the result of specific use-cases I’ve encountered in my own work. Building up a custom toolbox of useful code is rewarding, and a good goal-oriented framework for practicing whatever programming language you happen to be working in.